Quantitative Lattice Energy Analysis of Intermolecular Interactions in Crystal Structures of Some Benzimidazole Derivatives
Gopal Sharma and Rajni Kant* 
1Chemical Crystallography Laboratory, Department of Physics, University of Jammu, Jammu Tawi, India .
http://dx.doi.org/10.13005/OJPS05.01-02.08
Copy the following to cite this article:
Sharma G, Kant R. Quantitative Lattice Energy Analysis of Intermolecular Interactions in Crystal Structures of Some Benzimidazole Derivatives. Oriental Jornal of Physical Sciences 2020; 5(1,2). DOI:http://dx.doi.org/10.13005/OJPS05.01-02.08
Copy the following to cite this URL:
Sharma G, Kant R. Quantitative Lattice Energy Analysis of Intermolecular Interactions in Crystal Structures of Some Benzimidazole Derivatives. Oriental Jornal of Physical Sciences 2020; 5(1,2). Available From: https://bit.ly/3ECHRkm
Download article (pdf) Citation Manager Publish History



Introduction
Benzimidazole is an important class of heterocyclic aromatic organic compounds which consists of a benzene ring fused with imidazole ring. It is present in naturally biological active substances such as vitamin B12 and purine bases.1 It is an important scaffold beneficial for the development of pharmaceutically as well as biologically important molecules.2 Substituted benzimidazole derivatives have found diverse therapeutic applications such as anti-HIV,3 antiulcer,4 antihypertensive, antifungal, anthelmintic, antihistaminic and cardiotonic.5,6 Omeprazole and mebendazole are benzimidazole derivatives available in the market as proton pump inhibitors and as anthelmintics, respectively. The structure-activity relationship (SAR) studies suggest that substitution at C-2 position of this heterocyclic aromatic system highly influences the biological activity.6 Benzimidazole derivatives have been paid great heed because of their high pharmacological and biological activities. Their derivatives are one of the top frequently used ring systems for small molecule drugs listed by the US FDA.7
As a part of our ongoing research work on the preparation of X-ray diffraction quality single crystals and their structural analysis8,9, we have identified a series of five benzimidazole derivatives from CSD (version: 2020). The lattice and cohesive energies of all the molecular pairs were computed by using PIXEL10 software. The input CIF file required to carrying out the lattice energy calculations for each molecule was obtained from the CSD. The primary aim behind the analysis of lattice and intermolecular interaction energies is to compute and evaluate interaction energies which are associated with the molecular pairs and to investigate the contribution of these interactions in molecular packing stability11.The chemical structure of benzimidazole showing the numbering scheme is shown in Figure-1 and their derivatives are already reported with compound name, code, chemical structure are shown in Table-1.
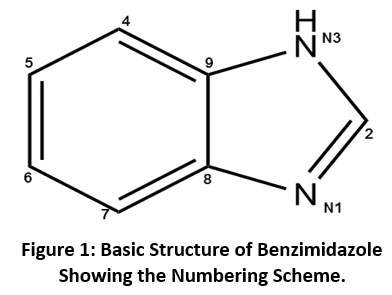 |
Figure 1: Basic Structure of Benzimidazole Showing the Numbering Scheme. Click here to view Figure |
Table 1: IUPAC Name and Chemical Structure of Benzimidazole Derivatives.
|
Code |
Compound IUPAC Name and References |
Chemical Structure |
|
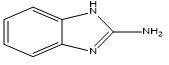
M-1 |
2-Aminobenzimidazole12 |
|
|
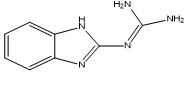
M-2 |
N''-1H-benzimidazol-2-ylguanidine13 |
|
|
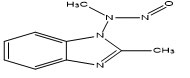
M-3 |
1-(N-Nitrosomethylamino)- |
|
|
M-4 |
2-nitro-1H-benzimidazole15 |
|
|
M-5 |
2-Chlorobenzimidazole16 |
|
Theoretical Calculations
Pixelc module in the CLP-PIXEL10 package (version 3.1 may 2016 available from http://www.angelogavezzotti.it) was performed to find out the intermolecular interaction and lattice energies in the crystal structures of benzimidazole derivatives as identified from the CSD. Hydrogen atom positions for the interaction energy calculations were assigned and intermolecular energies were determined based on numerical integrals over calculated electron densities of molecules computed by using GAUSSIAN0917 program. PIXEL allows for the total lattice energy to be divided into four main contributing terms: Coulombic, Polarization, Dispersion and Repulsion component. The sum of these four contributing terms gives the total interaction energy between the molecular pairs which assist to understand the role of intermolecular interactions in molecular packing.18-20 The total lattice energies for molecule M-1 to M-5 and their coulombic, polarization, dispersion and repulsion contributions are presented in Table-2 while the distance between centroids, symmetry code, the individual energy components and total energy between the molecular pairs are presented in Table-3. The total intermolecular interaction energies between the molecular pairs are arranged in the descending order and their pairs were examined using the Mercury21 software. The geometrical constraints put on the selected intermolecular pairs are the sum of van der Waals radius + 0.4Å and the directionality is greater than 110º.
Table 2: Lattice Energy for M-1 to M-5 (kcal/mol)
|
Molecule |
Coulomb Energy (ECoul) |
Polarization |
Dispersion |
Repulsive |
Total Energy |
|
M-1 |
- 22.87 |
- 10.92 |
- 24.19 |
24.81 |
- 33.17 |
|
M-2 |
- 27.82 |
- 16.13 |
- 34.11 |
36.09 |
- 41.97 |
|
M-3 |
- 9.92 |
- 3.68 |
- 23.97 |
13.98 |
- 23.59 |
|
M-4 |
- 19.48 |
- 9.46 |
- 26.31 |
26.24 |
- 29.13 |
|
M-5 |
- 17.57 |
- 8.39 |
- 27.17 |
24.16 |
- 28.97 |
Table 3: Interaction Energies (in kcal /mol) Between Molecular Pairs.
|
Mot-if |
Centroid |
ECoul |
EPol |
EDisp |
ERep |
ETot |
Symmetry |
Important |
|
M-1 |
||||||||
|
A |
7.188 |
-23.59 |
-10.16 |
- 6.12 |
21.37 |
- 18.52 |
1-x, -y, 1-z |
N2-H2A…N1 |
|
B |
4.476 |
-7.17 |
-2.70 |
-6.43 |
5.74 |
-10.56 |
-½+x, ½-y, z |
N3-H3…N1 |
|
C |
5.554 |
-2.32 |
-0.60 |
-2.99 |
0.96 |
-4.92 |
x, -1+y, z |
C7-N3 |
|
D |
7.700 |
-1.31 |
-1.67 |
-3.23 |
3.51 |
-2.70 |
½-x, -½+y, 1-z |
N2-H2B…N2 |
|
M-2 |
||||||||
|
A |
8.065 |
-20.15 |
-10.06 |
-8.05 |
23.83 |
-14.46 |
-x, 1-y, -z |
N4-H4A…N2 |
|
B |
5.068 |
-7.41 |
-3.30 |
-7.03 |
9.11 |
-8.63 |
½-x, -½+y, z |
C7-H7…π |
|
C |
5.241 |
-2.48 |
-1.15 |
-6.74 |
2.70 |
-7.67 |
-1+x, y, z |
N5-H5A…π |
|
D |
7.740 |
-3.03 |
-2.72 |
-5.52 |
5.59 |
-5.66 |
1/2 + x, 1/2 – y, -z |
N4-H4A…N5 |
|
E |
6.943 |
-1.46 |
-0.50 |
-1.84 |
0.45 |
-3.35 |
-1/2-x, -1/2+ y, z |
N5-H5A…N2 |
|
F |
7.622 |
-1.58 |
-0.79 |
-3.25 |
3.56 |
-2.05 |
3/2-x, -1/2+ y, z |
C4-H4…π |
|
G |
8.793 |
-0.60 |
-0.48 |
-3.18 |
2.29 |
-1.93 |
-1/2+x, y, 1/2-z |
C6-H6…π |
|
M-3 |
||||||||
|
A |
4.880 |
-0.21 |
-1.17 |
-10.18 |
5.52 |
-6.05 |
-x, 2-y, 1-z |
π…π |
|
B |
7.653 |
-3.80 |
-0.74 |
-3.51 |
2.46 |
-5.59 |
-x, 2-y, 2-z |
C10-H10C…N4 |
|
C |
6.866 |
-2.08 |
-0.57 |
-2.65 |
1.24 |
-4.06 |
1/2-x, 1/2+y, 3/2-z |
C10-H10A…π |
|
D |
6.890 |
-2.41 |
-0.88 |
-3.54 |
2.84 |
-3.99 |
-1/2-x, 1/2+y, 3/2-z |
C11-H11A…O1 |
|
E |
8.026 |
-1.46 |
-0.52 |
-3.39 |
2.01 |
-3.35 |
1/2+x, 3/2-y, 1/2+z |
C6-H6…N1 |
|
F |
6.930 |
-0.96 |
-0.84 |
-3.49 |
2.27 |
-2.99 |
1+x, y, z |
C10-H10B…N4 |
|
M-4 |
||||||||
|
A |
6.042 |
-13.77 |
-6.24 |
-6.48 |
13.29 |
-13.19 |
-1/4+x, 1/4-y, -1/4+z |
C4-H4…O2 |
|
B |
3.713 |
-0.17 |
-1.05 |
-9.87 |
7.46 |
-3.61 |
x, y, -1+z |
π…π |
|
C |
9.817 |
-1.77 |
-0.45 |
-1.79 |
1.41 |
-2.63 |
1/4-x, 1/4+y, |
C5-H5…O2 |
|
D |
7.563 |
-1.55 |
-0.36 |
-1.15 |
0.88 |
-2.17 |
1/4+x, 1/4-y, |
C7-H7…O1 |
|
E |
7.746 |
-0.55 |
-0.29 |
-2.22 |
1.27 |
-1.82 |
1/2-x, -y, -1/2+z |
C6-H6…π |
|
M-5 |
||||||||
|
A |
6.736 |
-13.24 |
-6.02 |
-6.72 |
12.93 |
-13.02 |
-1/2+x, 3/2-y, 1-z |
C4-H4…C1 |
|
B |
5.375 |
-2.75 |
-0.81 |
-5.28 |
3.08 |
-5.74 |
1/2-x, -1/2+y, z |
C2…C4 |
|
C |
4.037 |
-0.86 |
-1.12 |
-8.99 |
5.88 |
-5.09 |
1-x, 1-y, 1-z |
C6…C1 |
|
D |
6.423 |
-0.67 |
-0.45 |
-3.32 |
2.48 |
-1.98 |
-1/2+x, y, 3/2-z |
C7-H7…C5 |
Results and Discussion
M-1: 2-Aminobenzimidazole
Distinct molecular pairs of molecule M-1 (A-D) as obtained from the crystal structure involved in the formation of intermolecular interactions are shown in Figure-2. The maximum stability to the molecular structure occurs with N2-H2A…N1 intermolecular interaction (Motif A, Figure-2). The pairs of N2-H2A…N1 interaction link the molecules into centrosymmetric dimers in the crystal, forming R22 (8) ring motif having interaction energy of -18.52 kcal mol-1 with 60% contribution to the net stabilization from coulombic energy, 25% contribution from polarization energy and 15% from the dispersive energy, respectively. The next stabilized pair (Motif B, Figure 2) shows the presence of N-H…N (involving H3 and N1), N-H…π and C-H…π interactions. The C4-H4…π interaction that connects the molecules between a hydrogen atom (H4) and atom C6 and C7 of Cg1 ring (where Cg1 represents centre of gravity of benzene ring), whereas interaction N3-H3…π involving atom (H3) with atom C7 and C8 of Cg1 as shown in Figure-2 (Motif B) having interaction energy of -10.56 kcal mol-1 with a maximum contribution of -7.17 kcal mol-1 from coulombic component. The next stabilized motif (C) in molecule M-1 involves C…N interaction with total interaction energy of -4.92 kcal mol-1 (40 % contribution from coulombic energy, 50% dispersion energy and 10% from polarization energy to the net stabilization). The last stabilized pair (Motif D, Figure-2) in molecule M-1 involves N-H…N (H2B and N2) weak intermolecular hydrogen interaction with total stabilization energy of -2.70 kcal mol-1 with 52% of the maximum contribution from dispersive energy, 27% of polarization energy and 21% of coulombic energy contribution to net stabilization. The packing for M-1 shows the formation of molecular sheets in the ac plane (Figure-3).
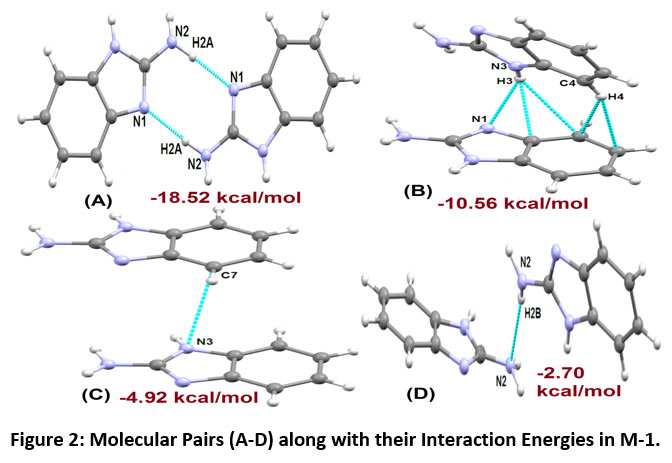 |
Figure 2: Molecular Pairs (A-D) along with their Interaction Energies in M-1. Click here to view Figure |
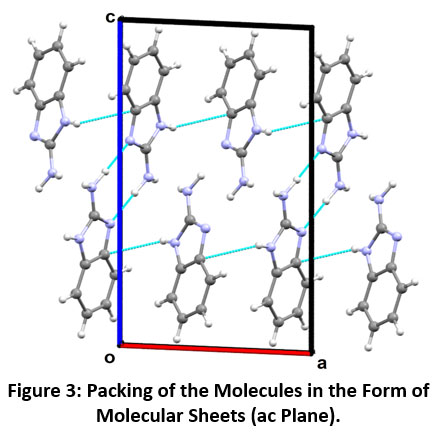 |
Figure 3: Packing of the Molecules in the Form of Molecular Sheets (ac Plane). Click here to view Figure |
M-2: N''-1H-benzimidazol-2-ylguanidine
The molecular pairs of compound M-2 and their respective interaction energies are shown in Figure-4. The contributed stabilization energy of the molecular pair (A) in compound M-2 is -14.46 kcal/mol (with 52 % major contribution from the coulombic component) is due to the presence of a N4-H4A…N2 hydrogen bond linking the molecules to form dimers (Figure-4 motif A). The motif (B), the second most stabilized pair, involves edge-to-face C-H…π interaction (involving H7 with C4 and C9 of Cg1) along with a weak intermolecular interaction (N3-H3…N1) having interaction energy -8.63 kcal/mol with 41 % maximum contribution from coulombic energy (Figure-4, Motif B). The motif (C) is the third most stabilized pair that show the presence of N5…Cg2 and N-H…π interactions (involving H5A and H5B with N1, C2, C8 and C9 respectively) contributing -7.67 kcal/mol energy towards stabilization. Motif (D) is the next most stabilized motif in the crystal packing involves N4-H4A…N5 and N5-H5A…N4 (atom N4 and N5 acts as a donor via H4A and H5A) bifurcated donor configuration having interaction energy of -5.66 kcal/mol with maximum contribution from dispersive energy. Motif (E) and (F) contributing -3.35 kcal/mol and -2.05 kcal/mol, showing the presence of N5-H5A…N2 and edge-to-face C-H…π interaction (involving H4 with C6 and C7 of Cg1 ring) with maximum contribution from dispersive energy. The molecular packing in M-2 showing the formation of sheets in the bc plane as shown in Figure-5.
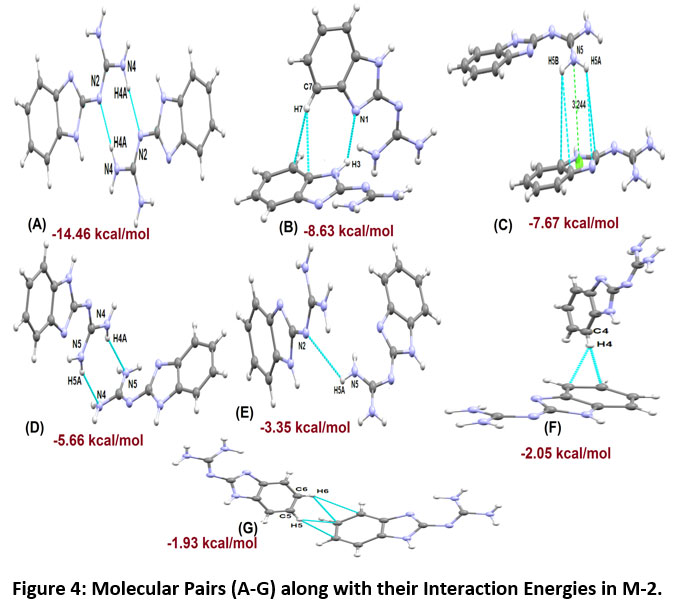 |
Figure 4: Molecular Pairs (A-G) along with their Interaction Energies in M-2. Click here to view Figure |
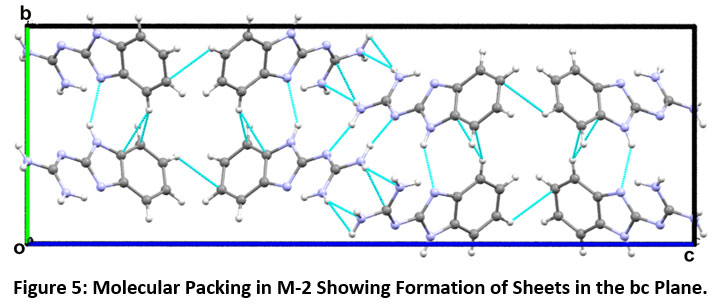 |
Figure 5: Molecular Packing in M-2 Showing Formation of Sheets in the bc Plane. Click here to view Figure |
M-3: 1-(N-Nitrosomethylamino)-2-methylbenzimidazole
The molecular pairs of M-3 extracted via crystal packing carrying their respective stabilization energies are shown in Figure-6. The motif (A) shows π…π stacking with total stabilization energy of -6.05 kcal/mol (88 % contribution from coulombic energy). In motif (B) exists C10-H10C…N4 interaction which links the pair of molecules to form an inversion dimer having interaction energy of - 5.59 kcal/mol. In motif (C), there exists an intermolecular interaction [C11-H11C…N1] and edge to face C-H…π stacking interaction (involving H10A interconnecting with C7 and C8 of Cg1 ring). This interaction contributes -4.06 kcal/mol energy towards stabilization. The molecular pair (D) shows the existence of C11-H11A…O1 and C-H…π interactions (involving H11B interconnecting with C4 and C5 of Cg1 ring), leading to the total interaction energy -3.99 kcal/mol with a large contribution from the dispersive energy component. Some other molecular pair (motif E) shows the existence of C5-H5…O1 and C-H…π interaction involving H6 interconnecting with C2 and N1 of Cg2 ring having total interaction energy - 3.35 kcal/mol and the stabilization largely come from a dispersive component. The least most stabilized pair (motif F) involves the existence of C-H…N type (C10-H10B…N4 & C4-H4…N1) intermolecular interactions with the total energy of -2.99 kcal/mol (dispersive energy 65% contribution). π...π interaction and C-H…N interaction link the molecules to form a molecular chain along with c- axis as shown in Figure-7.
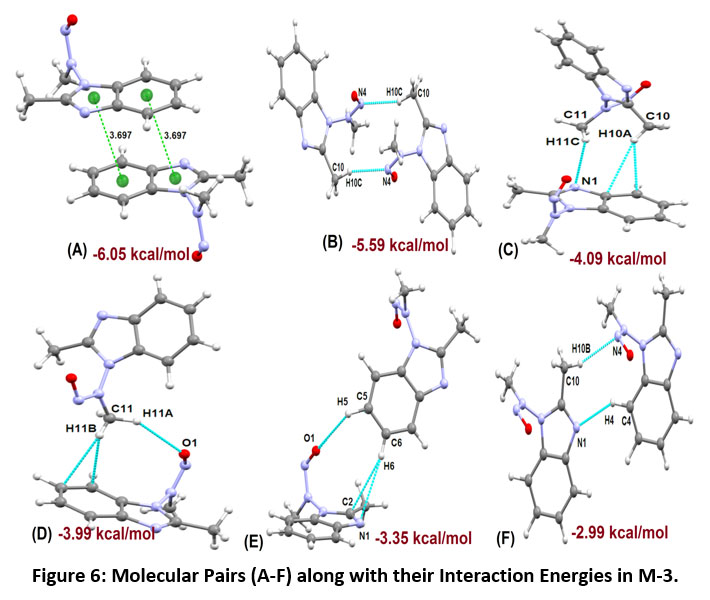 |
Figure 6: Molecular Pairs (A-F) along with their Interaction Energies in M-3. Click here to view Figure |
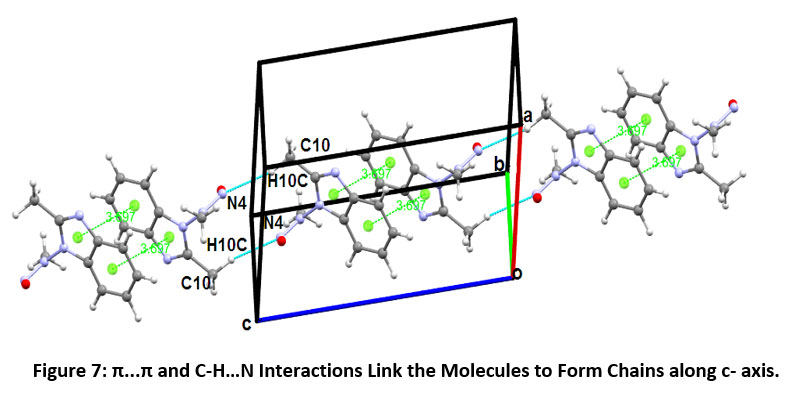 |
Figure 7: π...π and C-H…N Interactions Link the Molecules to Form Chains along c- axis. Click here to view Figure |
M-4: 2-nitro-1H-benzimidazole
The molecular pairs of 2-nitro-1H-benzimidazole (M-4) extracted via crystal packing carrying their corresponding stabilization energies are shown in Figure-8. The motif (A) in the molecule M-4 is the most stabilized pair that involves C4-H4…O2, N3-H3…N1 and C7-H7…O1 interactions having energy component of -13.19 kcal/mol ( maximum contribution from coulombic energy). The total stabilization energy (-3.61 kcal/mol) of the motif (B) is the second most stabilized pair indicating the presence of N2…C2, O1…C2, O1…N2 and π…π interactions(maximum contribution from the dispersive component).
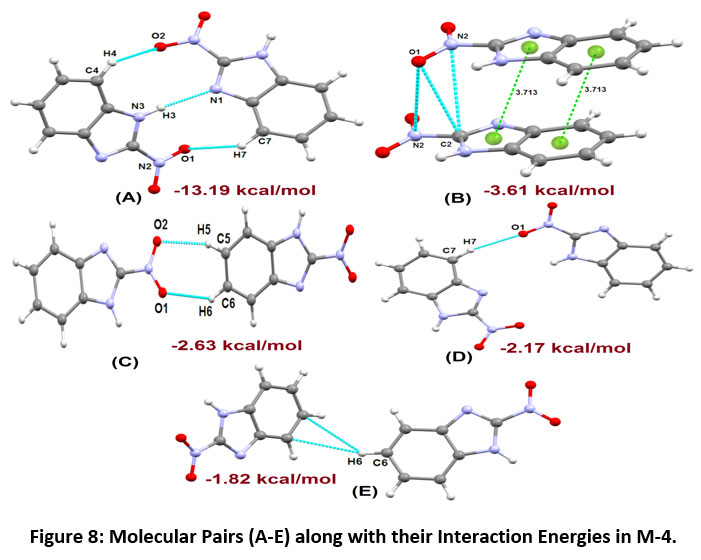 |
Figure 8: Molecular Pairs (A-E) along with their Interaction Energies in M-4. Click here to view Figure |
The Motif (C) has an interaction energy of -2.63 kcal/mol, indicating the presence of C-H…O intermolecular interactions (involving H5 with O2 and H6 with O1) with almost equal contribution from dispersive and coulombic component. The Motif D & E involves C7-H7…O1 and C-H…π (involving H6 with C6 and C7 of Cg1) interactions having -2.17 kcal/mol and -1.82 kcal/mol energy component, respectively. The packing in the crystal structure of 2-nitro-1H-benzimidazole M-4 indicates the formation of molecular sheets in the ab plane (Figure-9).
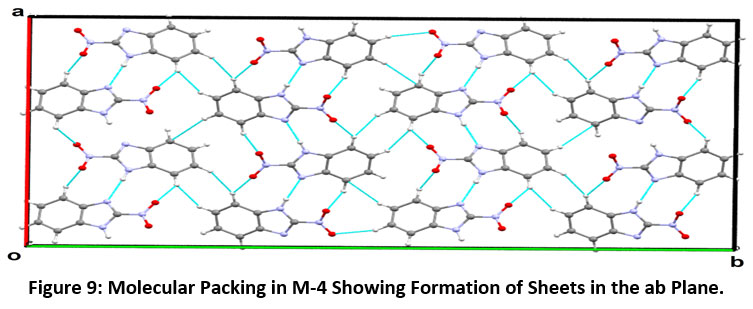 |
Figure 9: Molecular Packing in M-4 Showing Formation of Sheets in the ab Plane. Click here to view Figure |
M-5: 2-Chlorobenzimidazole
The stabilized pairs of 2-Chlorobenzimidazole (M-5) are shown in Figure-10. The Motif (A) is involved in the formation of C4-H4…Cl and N3-H3…N1 intermolecular interactions having total interaction energy of -13.02 kcal/mol. The pairs of N2-H2A…N1 interaction connect the molecules into inversion dimers in the crystal, generating R22 (12) ring motif. The interaction energy component of Motif B is - 5.74 kcal/mol, indicating the existence of C4…C2 and C4…N3 interactions (maximum contribution coming from the dispersive component).
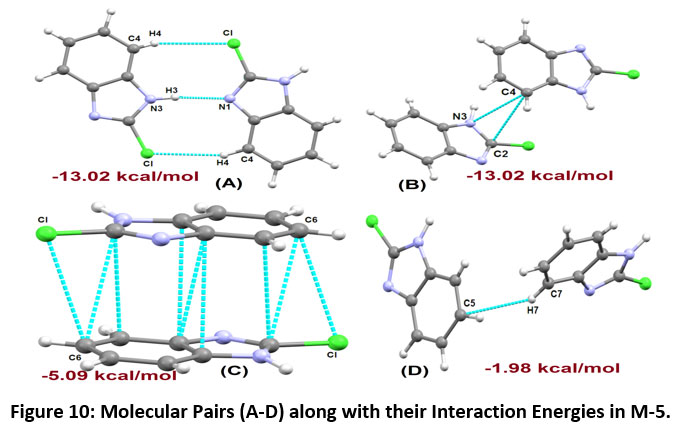 |
Figure 10: Molecular Pairs (A-D) along with their Interaction Energies in M-5. Click here to view Figure |
The Motif C, involved in the molecular stacking, has an interaction energy of - 5.09 kcal/mol (with 82 % contribution from dispersive energy towards the net stabilization). The last stabilized pair involves C7-H7…C5 having total interaction energy of -1.98 kcal/mol as shown in [Motif (D), Figure-12]. The molecular packing of compound M-5 is shown in Figure-11.
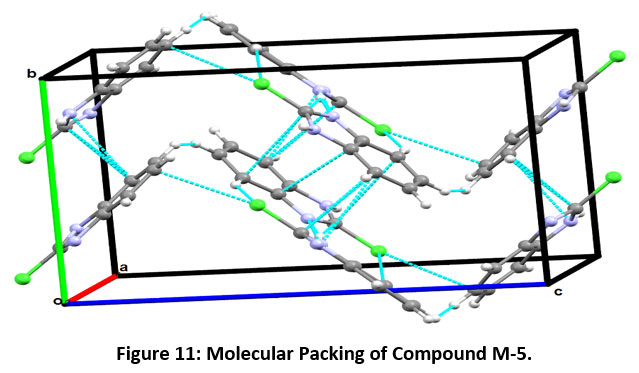 |
Figure 11: Molecular Packing of Compound M-5. Click here to view Figure |
Conclusions
- In case of all the five benzimidazole structures, the lattice energy lies in the range -23 to -42 kcal/mol and is separated into coulombic, polarization, dispersion and repulsion components with different energy contributions.
- The maximum contribution comes from the dispersion energy.
- The stability to the molecular structure comes from the molecular pairs interacting via intermolecular interactions (N-H…N and C-H…N).
- C-H…π and N-H…π type edge-to-face stacking interactions have also been found to be contributing substantially in the stabilization of their crystal structures.
- The pairs of N-H…N intermolecular hydrogen bonds, responsible for the formation of centrosymmetric dimmers (with an energy contribution of -14 to - 18.52 kcal/mol), lend credible support to molecular packing.
- The energy of molecular pairs interacting via C-H…N interactions link the molecules into dimers (energy range being -2 to -5 kcal/mol).
Lattice energy calculation is a useful method to assess the stability of crystal structures in which Coulombic and dispersion type interactions make up an essential part of the intermolecular interactions. The work reported in this paper shows the existence of different key structural motifs that assist stabilization of molecular packing in the unit cell. The calculation of interaction energy of the molecular pairs by PIXEL help us determine the strength of each interaction and the role played by the weak intermolecular interactions in molecular structure determination also gets confirmed. The study of these interactions helps design some new and more fascinating biologically active benzimidazole derivatives by changing the strength of donor and acceptor atoms.
Acknowledgements
Rajni Kant acknowledges the Research Grants as sanctioned under RUSA 2.0 Project (Ref. No: RUSA/JU/2/2019-20/111/3588-3636).
References
- Datta, A. and Roy, S., Solid Phase Synthesis of Biologically active Benzimidazole Derivatives Catalysed by CH3 SO3 H-SiO2 Under Solvent free Condition, Orient. J. Chem., 36(3), 537-543, (2020).
CrossRef - Arjmand F. and Aziz M., Synthesis and characterization of dinuclear macrocyclic cobalt (II), copper (II) and zinc (II) complexes derived from 2, 2, 2′, 2′-S, S [bis (bis-N, N-2-thiobenzimidazolyloxalato-1, 2-ethane)]: DNA binding and cleavage studies., Eur. J. Med. Chem., 44, 834-844, (2009).
CrossRef - Chimirri A., Grasso S., Monforte A. M., Monforte P., Zappala M., Anti-HIV agents. I: Synthesis and in vitro anti-HIV evaluation of novel 1H, 3H-thiazolo [3, 4-a] benzimidazoles, Farmaco, 46, 817-823, (1991).
- Ishihara K., Ichikawa T., Komuro Y., Ohara S. and Hotta K., Effect on gastric mucus of the proton pump inhibitor leminoprazole and its cytoprotective action against ethanol-induced gastric injury in rats, Arzneim. Forsch., 44, 827-830, (1994).
- Taylor R. D., MacCoss M. and Lawson A. D. G., Rings in drugs: Miniperspective, J. Med. Chem., 57, 5845-5859, (2014).
CrossRef - Rajasekhar S., Maiti B., Balamurali M. & Chanda K., Synthesis and medicinal applications of benzimidazoles: An overview, Curr. Org. Synth., 14(1), 40-60, (2017).
CrossRef - Marinescu M., (Ed.). Chemistry and applications of benzimidazole and its derivatives, BoD–Books on Demand, (2019).
CrossRef - Sharma, R., Samshuddin S., Narayana B., Gupta V. and Kant R., Styructural analysis of (2E)-3-[4-(1H-Benzimidazol-2-ylmethoxy)phenyl]-1-(4-chlorophenyl)prop-2-en-one, Eur. Chem. Bull., 3, 214-216, (2014).
- Yadav G. & Ganguly S., Structure activity relationship (SAR) study of benzimidazole scaffold for different biological activities: A mini-review, Eur. J. Med. Chem., 97, 419-443, (2015).
CrossRef - Gavezzotti A., Calculation of lattice energies of organic crystals: the PIXEL integration method in comparison with more traditional methods, Z. Krist., 220, 499-510, (2005).
CrossRef - Salian V. V., Narayana B., Byrappa K., Sarojini B. K. and Kant R., X-ray structure, hydrogen bonding and lattice energy analysis of (2 E)-1-(anthracen-9-yl)-3-(4-substitutedphenyl) prop-2-en-1-ones, Bull. Mater. Sci., 39, 1851-1860, (2016).
CrossRef - Wulff-molder, D., Meisel, M., CSD Comm. (1999).
- Skelton B. W., CSD Comm. (2017).
- Pozharskii A. F., Dyablo O. V., Belyaev A. V., Starikova Z. A. and Yanovsky A. I., Synthesis and some properties of 1-(N-Nitrosoalkylamino) benzimidazoles, Tetrahedron, 54, 9677-9688, (1998).
CrossRef - Klapotke T. M., Preimesser A. and Stierstorfer J., Energetic Derivatives of 2?Nitrimino?5, 6?dinitro?benzimidazole, Propellants, Explos., Pyrotech., 40, 60-66, (2015).
CrossRef - Panneerselvam K, Soriano-García M., 2-Chlorobenzimidazole, Acta Crystallogr., C 52,1799-1801, (1996).
CrossRef - Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Scalmani G., Barone V., Mennucci B., Petersson G. A., Nakatsuji H., Caricato M., Li X., Hratchian H. P., Izmaylov A. F., Bloino J., Zheng G., Sonnenberg J. L., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T., Montgomery J. A., Peralta J. E., Ogliaro F., Bearpark M., Heyd J. J., Brothers E., Kudin K. N., Staroverov V. N., Kobayashi R., Normand J., Raghavachari K., Rendell A., Burant J. C., Iyengar S. S., Tomasi J., Cossi M., Rega N., Millam N. J., Klene M., Knox J. E., Cross J. B., Bakken V., Adamo C., Jaramillo J., Gomperts R., Stratmann R. E., Yazyev O., Austin A. J., Cammi R., Pomelli C., Ochterski J. W., Martin R. L., Morokuma K., Zakrzewski V. G., Voth G. A., Salvador P., Dannenberg J. J., Dapprich S., Daniels A. D., Farkas O., Foresman J. B., Ortiz J. V., Cioslowski J. & Fox D. J., Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford, CT, (2016).Carlucci L. and Gavezzotti A., Molecular recognition and crystal energy landscapes: An X-ray and computational study of caffeine and other methylxanthines, Chem Eur J., 11, 271-279, (2005).
CrossRef - Carlucci L. and Gavezzotti A., Molecular recognition and crystal energy landscapes: An X-ray and computational study of caffeine and other methylxanthines, Chem Eur J., 11, 271-279, (2005).
CrossRef - Dunitz J. D., Gavezzotti A., Supramolecular synthons: Validation and ranking of intermolecular interaction energies, Cryst Growth Des., 12, 5873-5877, (2012).
CrossRef - Dunitz J. D. and Gavezzotti A., Toward a quantitative description of crystal packing in terms of molecular pairs: application to the hexamorphic crystal system, Cryst Growth Des., 5, 2180-2189, (2005).
CrossRef - Macrae C. F., Bruno I. J., Chisholm J. A., Edgington P. R., McCabe P., Pidcock E., Rodriguez-Monge L., Taylor R., van de Streek J. & Wood P. A., Mercury CSD 2.0-New Features for the Visualization and Investigation of Crystal Structures, J. Appl. Crystallogr., 41, 466-470, (2008).
CrossRef

This work is licensed under a Creative Commons Attribution 4.0 International License.
