Computational Design of Organic Laser Materials Based on Porphyrin Derivatives: A DFT and Optical Study
Talib Zeedan Al-Mosawi*
1Department of Laser and Optoelectronics Engineering, University of Kut, Kut, , 52002 Wasit, Iraq .
talib.almosawi@alkutcollege.edu.iq
http://dx.doi.org/10.13005/OJPS11.01.01
Porphyrin-based compounds have good electronic and optical properties, which can be beneficial for use in optoelectronics. In this paper, the electronic and optical properties of porphyrin, PAI, and PMg were studied using the DFT and TD-DFT methods. The HOMO-LUMO analysis showed that the energy gap decreased significantly for the modified compounds. Among the modified compounds, PMg showed the lowest energy gap and the highest flexibility. The DOS analysis showed that the density of states increased near the Fermi level for the modified compounds, especially PMg. The TD-DFT analysis showed that the transition energy for the porphyrin compound was weak, with ? = 489.27 nm and f = 0.0075. For the PMg compound, the transition energy was strong, with ? = 458.11 nm and f = 0.6480, which indicated a highly allowed transition. The results showed that the PMg compound performed well in the electronic and optical properties, which makes it a good candidate for use in optoelectronics and lasers.
Copy the following to cite this article:
Talib Zeedan Al-Mosawi. Computational Design of Organic Laser Materials Based on Porphyrin Derivatives: A DFT and Optical Study. Oriental Jornal of Physical Sciences 2026; 11(1)
DOI:http://dx.doi.org/10.13005/OJPS11.01.01Copy the following to cite this URL:
Talib Zeedan Al-Mosawi. Computational Design of Organic Laser Materials Based on Porphyrin Derivatives: A DFT and Optical Study. Oriental Jornal of Physical Sciences 2026; 11(1)
Download article (pdf) Citation Manager Publish History



Introduction
Notably, structures based on porphyrins have shown promising results over the last few years owing to their electronic structure and photophysical properties. Porphyrins have shown a highly delocalized structure that enables efficient electron transfer and light-matter interactions. Such properties of porphyrins have shown promising results for potential application in the development of optoelectronic devices, solar cells, sensors, and laser-based structures. Furthermore, the potential for modifying the electronic structure of porphyrins has shown promising results for the development of advanced functional structures with unique optical properties [1-2].
One of the most promising strategies for enhancing the performance of porphyrin-based structures is the incorporation of metal ions within the core of the macrocyclic structure. Such modifications have shown promising results for enhancing the electronic structure of the porphyrin-based structures by reducing the HOMO-LUMO bandgap. Furthermore, the incorporation of metal ions within the porphyrin-based structures has also shown promising results for enhancing the electronic state of the structures. Such modifications are critical for assessing the optical absorption characteristics of the structures, which are critical for developing photonic and laser-based structures [3-6].
In this regard, various computational techniques such as density functional theory (DFT) and time-dependent DFT (TD-DFT) have shown promising results for playing a critical role in the understanding of the electronic and optical properties of molecular structures. DFT calculations have shown promising results for accurately determining the frontier molecular orbitals and global reactivity descriptors of molecules. Furthermore, TD-DFT calculations have shown promising results for accurately determining the electronic properties of molecules. Moreover, density of states calculations has shown promising results for a precise evaluation of the electronic properties of molecules by analyzing the molecular orbitals of the molecules [7-8].
In the present work, a systematic analysis has been carried out to understand the effect of structural modification on the electronic and optical properties of the porphyrin-based compounds. For the purpose of the present work, three structures of the porphyrin-based compounds were considered for the analysis: pristine porphyrin, PAI, and PMg. DFT and TD-DFT calculations were carried out to understand the frontier molecular orbitals of the compounds, global reactivity descriptors of the compounds, orbital distribution of the compounds, density of states of the compounds, and UV-Vis absorption characteristics of the compounds. Emphasis was placed on understanding the relationship between the electronic properties of the compounds and their optical properties [9-10]. From the results obtained in the present work, it is clearly understood that structural modification plays a critical role in determining the electronic properties of the porphyrin-based compounds. For instance, it is clearly understood from the present work that the addition of magnesium to the porphyrin-based compounds plays a critical role in increasing the electronic excitation probability of the compounds and enables the compounds to be used for optical and related applications such as lasers.
Computational Methods
In all quantum chemical computations, the Gaussian 16 program is employed. In the quantum chemical computations of the molecular structure of porphyrin, PAI, and PMg, the DFT method is employed to fully optimize the molecular structures of the compounds without any symmetry constraint [11-12]. In the quantum chemical computations of the molecular structures of the compounds, the B3LYP functional is employed. In addition, the 6-31G(d,p) basis set is employed for all the atoms of the molecules. Furthermore, the effective core potential is employed for all the metal atoms of the molecules. Frequency computations are carried out for all the optimized structures of the compounds by the same method as before to prove that all the optimized structures are true minima on the potential energy surface of the molecule by not having imaginary frequency values on their IR spectra, which are calculated during the optimization process. Furthermore, the calculation of the frontier molecular orbitals of the compounds is done to obtain the values of the HOMO and LUMO orbitals of the molecules. Using the values of the HOMO and LUMO orbitals of the molecules, some global descriptors of reactivity are calculated. These global descriptors of reactivity are IP, EA, electronegativity, chemical potential, global hardness, softness, and electrophilicity values by using the values of the HOMO and LUMO orbitals of the molecules by Koopmans' theorem.
The electronic excitation properties have also been calculated using the time-dependent density functional theory method. The calculations have been carried out using the same level of theory used for the calculation of the compounds' ground state. The lowest excited singlet state has been calculated for the compounds under investigation. The excitation energy, wavelength, and oscillator strength have also been calculated for the compounds. The results obtained have been used to calculate the UV-Vis spectrum of the compounds under investigation. In order to achieve a deeper understanding of the electronic properties of the compounds under investigation, the density of states calculation has been carried out using the Materials Studio program. The DOS plots have been calculated for the compounds under investigation. The analysis of the electronic state distribution has also been carried out for the compounds under investigation. The effect of the modification of the structure on the electronic density at the Fermi level has also been understood for the compounds under investigation. In all the calculations carried out in this work, GaussView has been used for the visualization of the molecular orbitals and the electronic distribution of the compounds under investigation. The results obtained have shown that they will be used for the analysis of the electronic properties of the compounds under investigation. The calculations carried out in this work have enabled a detailed understanding of the electronic properties of the compounds under investigation.
Results and discussion
Global Reactivity Analysis
The electronic structure of the aforementioned systems was examined based on the FMO theory, while the aforementioned theory refers to the use of the energy levels of the HOMO and LUMO orbitals for the calculation of some of the most important global descriptors of the chemical reactivity of the system, i.e., IP, EA, electronegativity, chemical potential, global hardness, softness, and electrophilicity [13-15].
Based on the electronic structure of the aforementioned system, the following conclusions can be drawn: The HOMO level is the lowest, i.e., the energy level of the HOMO orbital is the lowest, equal to -0.26955 a.u., while the energy level of the LUMO orbital is relatively deep, equal to -0.03633 a.u. As a result, the energy gap between the aforementioned orbitals is large, i.e., ?E = 0.23322 a.u. Therefore, it is concluded that the aforementioned system possesses the highest IP = 0.26955 a.u. and electronegativity = 0.15294 a.u., thus having a strong tendency to maintain the electronic density of the system and avoid any possible electron transfer. Additionally, the high value of global hardness, i.e., ? = 0.11661 a.u., and the relatively low value of softness, i.e., S = 4.2878 a.u., of the aforementioned system indicate the high electronic stability of the aforementioned system and the low chemical reactivity of the system. Finally, the relatively high value of the electrophilicity index, i.e., ? = 0.1003 a.u., of the aforementioned system indicates the moderate ability of the aforementioned system to attract an electron under appropriate conditions (Table 1). On the contrary, PAI has a high energy level of HOMO equal to -0.07731 a.u., a positive energy level of LUMO equal to 0.03136 a.u., and a large decrement in the bandgap ?E = 0.10867 a.u. The decrement in the bandgap is related to the high values of the electronic flexibilities and the high degree of susceptibility to the charge redistribution of electrons. The low ionization potential of PAI, equal to 0.07731 a.u., and low negative affinity of PAI, equal to -0.03136 a.u., indicate a low degree of stability related to the additional electron. The low electronegativity of PAI, equal to 0.02298 a.u., indicates a low degree of attraction related to electrons. The low hardness of PAI, equal to 0.05434 a.u., and high softness of PAI, equal to 9.2022 a.u., indicate a high degree of reactivity and electronic polarization related to the porphyrin system. The low electrophilicity index of PAI, equal to 0.0049 a.u., indicates a low degree of PAI system activity related to the acceptance of electrons. Likewise, the electronic properties of the PMg system were found to be intermediate between the electronic properties of the porphyrin and PAI systems. The HOMO level of the PMg system was found to be -0.11866 a.u., while the LUMO level of the PMg system was found to be -0.01244 a.u. This resulted in the lowest band gap energy among the studied systems, i.e., ?E = 0.10622 a.u., indicating the highest degree of electronic delocalization. The IP of the PMg system was found to be 0.11866 a.u., while the EA of the PMg system was found to be 0.01244 a.u., indicating the balanced ability of the PMg system to donate and accept electrons. The PMg system was found to possess the lowest hardness, i.e., ? = 0.05311 a.u., and the highest softness, i.e., S = 9.4144 a.u., indicating the highest degree of electronic flexibility and reactivity. The value of the ? parameter of the PMg system was found to be 0.0405 a.u., which is greater than the PAI system and less than the porphyrin system. The order of electronic reactivity of the studied compounds is as follows:
PMg > PAI > Porphyrin,
where PMg is the most reactive and electronically soft compound, and porphyrin is the most stable and hardest compound. From the results obtained in the present work, it is evident that the inclusion of a metal has a significant impact in controlling the electronic properties of the porphyrin structure, thus allowing a certain degree of control over the reactivity and stability of the compound by merely adjusting the energies of the frontier orbitals.
Table 1. Computed frontier molecular orbital energies and global reactivity descriptors for the Porphyrin, PAI, and PMg.
Comp. | EHOMO (a.u.) | ELUMO (a.u.) | ?E (a.u.) | IP | EA | ? | ? | ? | S | ? |
Porphyrin | -0.26955 | -0.03633 | 0.23322 | 0.26955 | 0.03633 | 0.15294 | -0.15294 | 0.11661 | 4.2878 | 0.1003 |
PAI | -0.07731 | 0.03136 | 0.10867 | 0.07731 | -0.03136 | 0.02298 | -0.02298 | 0.05434 | 9.2022 | 0.0049 |
PMg | -0.11866 | -0.01244 | 0.10622 | 0.11866 | 0.01244 | 0.06555 | -0.06555 | 0.05311 | 9.4144 | 0.0405 |
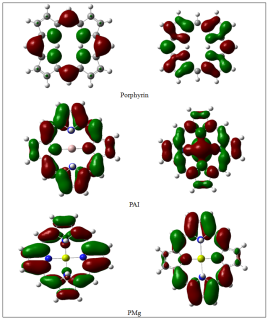
Frontier Molecular Orbital Distribution Analysis
The spatial distribution of the frontier molecular orbitals also provides additional information concerning the electronic properties of the investigated compounds, apart from the energy level data. As illustrated in Figure 1, for the pristine compound, it is observed that the densities of both the HOMO and LUMO molecular orbitals are delocalized across the whole conjugated structure of the macrocycle. The delocalization of ?-electrons across the whole structure is a characteristic feature of a highly symmetric electronic structure, where both molecular orbitals are equally distributed across the whole porphyrin ring [16-17].
In the PAI system, significant differences are observed concerning the spatial distribution of both the HOMO and LUMO molecular orbital densities. Specifically, whereas the HOMO density is partially localized across specific areas of the macrocycle, the LUMO density is also asymmetrically distributed across the whole structure of the macrocycle. The presence of "hot spots" of a higher concentration of electrons indicates a higher degree of electronic polarization for the PAI system, as evidenced by the calculated descriptors of the energy gap and softness.
However, for the case of PMg, a more significant change in the electron density distribution is observed. Thus, for the case of PMg, it was found that the HOMO is localized in the coordination area of the central metal and the adjacent nitrogen atoms, while the LUMO is localized in different parts of the macrocycle. Such a high degree of delocalization and the flow of electrons in a specific direction contribute to a decrease in the band gap and an increase in reactivity.
Analysis of the distribution of the molecular orbitals of the studied systems has shown the appropriateness of the previously obtained electronic descriptors. The change in the electronic structure of the porphyrin macrocycle in the presence of a metal atom has been proved. The transition from a completely delocalized state to a more polarized state is characteristic of the PAI and PMg systems. Among the studied systems, PMg is the most suitable for the flow of charges due to the high degree of localization of the HOMO and LUMO orbitals and the high degree of electronic polarization.
Density of States (DOS) Analysis
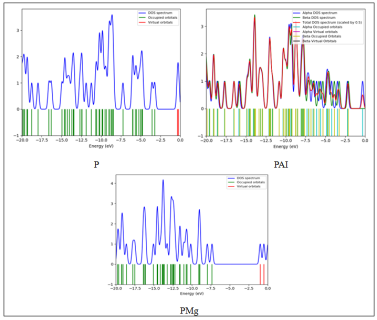
The electronic structure of the studied systems was also investigated through density of states analysis, in order to obtain a complete understanding of the results. Based on the above, it is obvious that, as depicted in Figure 2, the DOS plots indicate the differences in the electronic structure of the Porphyrin, PAI, and PMg systems [18].
In the case of the pristine Porphyrin system, it is obvious that the DOS plot indicates the presence of a relatively well-defined energy gap around the Fermi level, along with the relatively clear distinction between the occupied and virtual orbitals. The occupied orbitals are packed relatively densely in the region of the energy distribution below -8 eV, while the virtual orbitals exist above the energy gap, presenting relatively small overlap. The above electronic structure reveals the presence of a relatively stable configuration, along with the relatively small number of accessible states. The above statement is verified based on the relatively high HOMO-LUMO gap and the chemical hardness of the Porphyrin molecule.
In the case of the PAI system, it is obvious that the energy distribution is relatively compressed, especially in comparison to the Porphyrin molecule. In the region around the frontier of the energy distribution, it is obvious that the relatively small energy gap exists between the occupied and virtual orbitals. The DOS plots indicate the presence of relatively high peaks around the Fermi level, along with the relatively small distances between them. The above statement reveals the presence of a relatively high density of accessible states. In addition, it is obvious that the DOS plots for the alpha and beta spins are relatively similar to each other, indicating the absence of magnetic contributions in the system under investigation. Similarily, for the PMg system, the band gap is reduced, as the peaks are closer to each other on the DOS plot compared to the porphyrin system. Regarding the occupied and unoccupied states, it is obvious that a degree of convergence occurs between them. Moreover, from the figure 2, it is obvious that the intensity of the peaks is high for the PMg system compared to the porphyrin system. This is an indication of a degree of improvement observed in the electronic properties of the system. From the DOS plot of the porphyrin-based systems, it is obvious that the metal has a crucial role to play in modifying the electronic properties of the porphyrin-based system by increasing the density of states in the frontier region and by reducing the band gap between the HOMO and LUMO orbitals. The electronic accessibility of the porphyrin-based systems is as follows:
PMg ? PAI >> Porphyrin
Both systems have been able to display improved electronic properties and reactivity compared to the original porphyrin-based system.
Optical Transition Analysis
The TD-DFT results presented in Table 2 clearly indicate the differences in the electronic excitation characteristics for the studied systems. In the case of the pristine porphyrin molecule, the results clearly indicate the presence of the main absorption band at 489.27 nm with an extremely low oscillator strength (f = 0.0075). The low oscillator strength clearly indicates the weak allowed nature of the ? ? ?* transition, showing low probability for the electronic excitation process [19-20]. Such low probability might be related to the large HOMO-LUMO gap of the porphyrin molecule.
For the PAI molecule, the presence of the main absorption band at around 500 nm with almost the same oscillator strength, i.e., around 0.007, clearly shows the slightly improved probability for the electronic excitation process. Although some improvement has been made by modifying the structure of the porphyrin molecule, the overall results for the PAI molecule clearly indicate the low to moderate probability for the electronic excitation process.
The results for the PMg molecule clearly indicate the improved probability for the electronic excitation process by showing the high oscillator strength for the ? ? ?* transition, i.e., f = 0.6480, for the main absorption band at 458.11 nm. Such improved probability for the electronic excitation process clearly shows the high efficiency of the PMg molecule for the electronic excitation process (Figure 3).
The order of the oscillator strength clearly shows the relative efficiency of the electronic excitation process for the studied molecules, i.e., PMg > PAI > Porphyrin, indicating the importance of the metal incorporation for the porphyrin molecule, making it the most efficient molecule for the electronic excitation process.
Table 2: Optical properties of Porphyrin, PAI, and PMg
Comp. | ?max (nm) | Energy (eV) | fmax | Nature of transition | Remark |
Porphyrin | 489.27 | 2.5341 | 0.0075 | ? ? ?* | weak transition |
PAI | ~500 | ~2.48 | ~0.007 | ? ? ?* | weak–moderate |
PMg | 458.11 | 2.7064 | 0.6480 | ? ? ?* | strong transition |
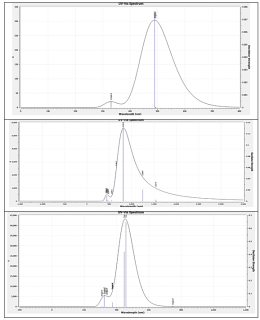 | Figure 3: Simulated UV–Vis spectrum of the Porphyrin, PAI, and PMg obtained from TD-DFT calculations
|
Conclusion
In the present research work, the electronic and optical properties of the porphyrin, PAI, and PMg compounds have been studied by using the DFT and TD-DFT methods. The results obtained from the present research work show that the structural modifications have a significant impact on the electronic and optical properties of the porphyrin structure. The HOMO-LUMO analysis of the compounds was also performed, and it was observed that the HOMO-LUMO gap of the compounds is decreased compared to the porphyrin structure. Among the compounds studied in the present research work, it was observed that the PMg compound has the least HOMO-LUMO gap, i.e., the compound is highly electronically flexible. The DOS analysis of the compounds was also performed, and it was observed that the density of the electronic states of the compounds is higher compared to the porphyrin structure. The TD-DFT analysis of the compounds was also performed, and it was observed that the porphyrin structure has a weak optical transition with very low intensity, while the PMg compound has a strong optical transition with high intensity in the visible region of the electromagnetic spectrum at 458.11 nm, i.e., 0.6480. The PAI compound showed intermediate results; i.e., the compound showed improvement in the intensity of the optical transition. It was concluded from the present research work that the incorporation of the metals has a significant impact on the electronic and optical properties of the porphyrin structure. Among the compounds studied in the present research work, it was observed that the PMg compound is highly effective for optoelectronic and laser devices.
Acknowledgements:
The authors sincerely acknowledge the University of Kut, for providing the necessary facilities and support to conduct this research.
Funding: The author received no specific funding for this research.
Conflicts of Interest/Competing Interests: The author declare no competing financial or non-financial interests.
Ethics Approval: Not applicable.
Clinical trial number: Not applicable
Consent to Participate: Not applicable.
Consent for Publication: The author has approved the manuscript for publication.
Data and/or Code Availability: The datasets generated and analyzed during this study are available from the corresponding author upon reasonable request.
References
1. Felix, I.M., Andrade, F.M., Woellner, C.F., Galvão, D.S., Tromer, R.M., 2024. Tuning the electronic and optical properties of two-dimensional diboron-porphyrin by strain engineering: A density functional theory investigation. Chemical Physics Letters 856, 141604.
2. El-Khalafy, S.H., Hassanein, M.T., Alaskary, M.M., Ramzy, G.H., Ali, A.I., 2025. Synthesis, characterization, and dielectric properties of bentonite clay modified with (3-chloropropyl)triethoxysilane and Co(II) porphyrin complex for technological and electronic device applications. Materials Advances 6, 1931–1949.
3. Hanna, L., Movsesian, E., Orozco, M., Bernot Jr., A.R., Asadinamin, M., Shenje, L., Ullrich, S., Zhao, Y., Marshall, N., Weeks, J.A., Thomas, M.B., Teprovich Jr., J.A., Ward, P.A., 2022. Spectroscopic investigation of the electronic and excited state properties of para-substituted tetraphenyl porphyrins and their electrochemically generated ions. Spectrochimica Acta Part A 278, 121300.
4. Ibrahim, M.A.A., Moussa, N.A.M., Rady, A.S.M., Mekhemer, G.A.H., El-Tayeb, M.A., Khan, S., Soliman, M.E.S., 2025. On the sensitivity of pristine and alkaline earth metal-decorated porphyrin-like porous C24N24 fullerenes toward dichlorosilane toxic gas: A DFT study. Chemical Physics 591, 112582.
5. Kumar, A., Al-Bahrani, M., Hossain, M.A., Mehedi, I.M., Iskanderani, A.I.M., Orosco Gavilán, J.C., Ramaiah, G.B., 2023. Transition metal-functionalized porphyrin-like C70 fullerenes as sensors and adsorbents of formaldehyde: DFT, NBO, and QTAIM study. Inorganic Chemistry Communications 153, 110883.
6. Esrafili, M.D., Kadri, M., 2023. Efficient delivery of anticancer 5-fluorouracil drug by alkaline earth metal functionalized porphyrin-like porous fullerenes: A DFT study. Journal of Molecular Graphics and Modelling 120, 108403.
7. Iqbal, M.M.A., Hussain, F., Javeed, H., Hassan, T., Hussain, R., 2026. Effect of ?-spacer moieties coupled to a porphyrin/PC70BM donor–acceptor for promising organic photovoltaic properties: a DFT study. RSC Advances 16, 16399–16417.
8. (No authors listed), 2026. Bis(4-nitroaniline) cobalt(II) meso-tetrakis(3,4,5-trimethoxyphenyl)porphyrin complex: spectroscopic, electrochemical and X-ray molecular structure characterizations, DFT/TD-DFT/MEP and NCI-RDG investigations and photodecolorization of the methyl Orange dye. Inorganica Chimica Acta 596, 123123.
9. Chen, L., Fu, L., Guan, Z., Liu, F., Huang, Z., Humphrey, M.G., Zhang, C., 2026. Ultrafast optical nonlinearity in porphyrin-based covalent organic framework membrane via Cu(II)-mediated interfacial polymerization. Materials Today Physics 62, 101967.
10. Machado, E.Q., Salles, B.B.E., Hantao, L.W., de Oliveira, C.J.F., Arruda, M.A.Z., 2026. Paper plate-assisted two-step fractionation and speciation for the evaluation of nickel and vanadium porphyrins via laser ablation inductively coupled plasma mass spectrometry. Talanta 298, 128988.
11. Takeda, K., Matsuo, M., Morita, M., & Yamamoto, T. (2023). Exploring TM–PICNC Systems: Adsorption on Metal Porphyrin Induced Carbon Nanocone. Frontiers in Chemistry, 11, 1132654.
12. Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., Scalmani, G., Barone, V., Petersson, G. A., Nakatsuji, H., Li, X., Caricato, M., Marenich, A. V., Bloino, J., Janesko, B. G., Gomperts, R., Mennucci, B., Hratchian, H. P., Ortiz, J. V., Izmaylov, A. F., Sonnenberg, J. L., Williams, C. F., Ding, F., Lipparini, F., Egidi, F., Goings, J., Peng, B., Petrone, A., Henderson, T., Ranasinghe, D., Zakrzewski, V. G., Gao, J., Rega, N., Zheng, G., Liang, W., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Vreven, T., Throssell, K., Montgomery Jr., J. A., Peralta, J. E., Ogliaro, F., Bearpark, M. J., Heyd, J. J., Brothers, E. N., Kudin, K. N., Staroverov, V. N., Keith, T. A., Kobayashi, R., Normand, J., Raghavachari, K., Rendell, A. P., Burant, J. C., Iyengar, S. S., Tomasi, J., Cossi, M., Millam, J. M., Klene, M., Adamo, C., Cammi, R., Ochterski, J. W., Martin, R. L., Morokuma, K., Farkas, O., Foresman, J. B., & Fox, D. J. (2016). Gaussian 16 Revision C.01. Gaussian, Inc., Wallingford CT.
13. Andreeva, N. A., & Chaban, V. V. (2018). Electronic and thermodynamic properties of the amino- and carboxamido-functionalized C60-based fullerenes: Towards non-volatile carbon dioxide scavengers. Journal of Chemical Thermodynamics, 116, 1–6.
14. Yoon, H. J., Lee, H., Kim, H., Park, J., Kim, J., & Lee, S. H. (2019). Recent advances in functionalized fullerenes for gas sensor applications. Journal of Materials Chemistry C, 7, 2603–2613.
15. Andreeva, N. A., & Chaban, V. V. (2018). Electronic and thermodynamic properties of the amino- and carboxamido-functionalized C60-based fullerenes: Towards non-volatile carbon dioxide scavengers. Journal of Chemical Thermodynamics, 116, 1–6.
16. Meelua, W., Wanjai, T., & Jitonnom, J. (2025). Exploring the effect of Zr/B ratio on the stability and reactivity of activated ?-caprolactone complexes: A DFT, QTAIM and NCI study. Journal of Molecular Graphics and Modelling, 136, 108960.
17. Reji, R. P., Sivalingam, Y., Kawazoe, Y., & Jayaraman, S. V. (2024). A quantum chemical assessment on the sensing ability of porphyrins and phthalocyanines towards volatile organic compounds using density functional theory investigations. Molecular Systems Design & Engineering, 9, 286–299.
18. Khaled, A., Kadri, R., & Berredjem, M. (2022). New Cu(II) and Zn(II) complexes with diethyl phenyl (N-phenylsulfamoylamino) methyl phosphonate: Synthesis, characterisation, DFT/M11 studies, NBO, DOS, QTAIM and RDG analysis. Journal of Molecular Structure, 1263, 133169.
19. Dahri, S., Messous, M.Y., Ahl Laamara, R., 2026. Mn-doped MgFeO? for UV–visible optoelectronics: A combined experimental and DFT study. Opt. Mater., January 2026.
20. Figueroa-Ariza, L.-T., Paez-Sierra, B.-A., 2026. Solvent-dependent spectral deconvolution of amino-substituted chalcones: UV–vis and FT-IR analysis supported by TD-DFT calculations. J. Mol. Struct., 5 February 2026 (Open Access).

This work is licensed under a Creative Commons Attribution 4.0 International License.